とある准教授の2025年の年収
とある准教授2年目の人間の年収です。 tonets.hatenablog.com
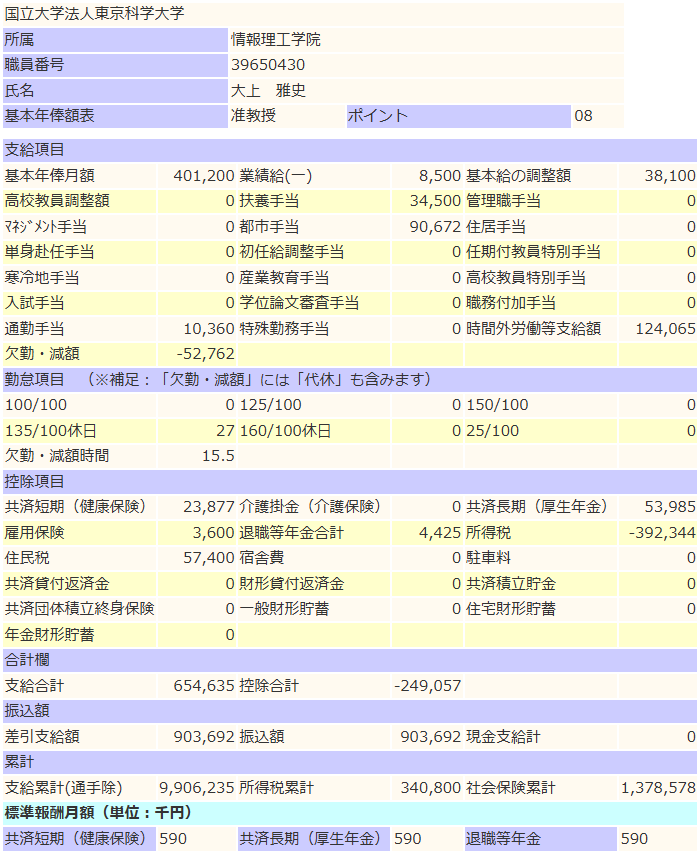
前年度比
- 支給額 +50,000円
- 所得税 +35,000円
- 社保 +30,000円
結果、収入は▲15,000円でした。
AlphaFoldは創薬の役に立つのか?
A. 役に立っているはず、たぶん。

はじめに
この記事は、創薬 (dry) Advent Calendar 2025 の16日目(大遅刻)の記事である。これまでに表題のような(煽りのような?)タイトルの講演を何度かしてきたが、その内容を噛み砕きながら文章化した。雰囲気が伝わるように感想レベルの話も(敢えて)混ぜているので、詳細は文献等をあたってほしい。
AlphaFoldとは?
AlphaFoldは、タンパク質の立体構造をアミノ酸配列から高精度に予測するAI技術/ソフトウェアである。2024年のノーベル化学賞の受賞テーマとしてもまだギリギリ記憶に新しく、AI企業であるGoogle DeepMindが開発したことでも話題になった。私の感覚では、進化関係を気軽に推測できるようにしたBLAST(配列アラインメント)に次ぐ画期的な成果であり、AlphaFoldによってタンパク質の形を気軽に推測できるようになったことは生命科学のあらゆる分野に大きな変化を与えたように思う。もちろんそれまでもタンパク質の立体構造予測は1つの予測タスクとして長く研究が続けられていたし、今もなお構造予測精度のさらなる改善を目指した研究が続いている(BLAST登場後にも数多くの配列アラインメントツールが発表されているのと同様である)。だが、AlphaFold(特にAlphaFold2)が成し得た精度は金字塔であり、そのインパクトは絶大であった。 www.jstage.jst.go.jp
タンパク質立体構造と創薬の関係
タンパク質の立体構造の情報は、創薬の役に立つとよく言われる。たとえばタンパク質の活性部位などのポケット(構造上の窪み)に対し、ぴったりハマるように念入りに設計された化合物を結合させてやれば、そのタンパク質の機能を変えさせることができそうである。もしタンパク質が疾病に関係しているものであれば、こうしてできた化合物は薬の候補になり得るだろう。タンパク質は、配列のみだとどんなアミノ酸が並んでいるかしかわからないが、立体構造があればポケットがどんなアミノ酸で構成されているか、どんな配置か、アミノ酸の側鎖がどんな向きになっているか、などなど一目瞭然である。立体構造にぴったりハマるように化合物を設計することが「合理的設計」(rational design) とか「合理的創薬」(rational drug discovery) と言われるように、立体構造によってタンパク質を狙った新しい薬の設計が可能になるわけである。ということで、タンパク質の立体構造の情報は、あると創薬の役に立ちそうである。
実際には、立体構造にハマる化合物を手作業で設計するのは大変なので、最初にたくさんの候補化合物を用意しておいて、ポケットにぴったりな形をしてそうなヤツをコンピューターで選んでおくことが多く、これを(構造ベースの)バーチャルスクリーニングと呼ぶ。だいたい数百万から、最近では数億化合物くらいの候補を準備しておいて、その中から絞り込んでいく計算をする。計算にはドッキングシミュレーションという方法が活用されることが多く、Schrödinger社のGlideというソフトウェアが製薬企業などでも比較的よく使われていると思う。 www.schrodinger.com
AlphaFold2と低分子化合物の合理的創薬
立体構造はあると便利そうだが、立体構造を解くのは実験の話になるのでコストがかかる*1。であれば、タンパク質の立体構造を正確に予測できるAlphaFoldは、まだ立体構造が解かれていないタンパク質に対して化合物を設計するのに役立ちそうである。
実際、多くの研究者がそのように考えて、試した結果が論文化されている。AlphaFold2で予測した立体構造で、どのくらい構造ベースのバーチャルスクリーニングがうまくいくかを、すでに答えを知っている活性化合物/非活性化合物たちで評価して答え合わせをするわけである(良し悪しは、答え合わせ時の点数に相当するROC-AUCやEnrichment Factorといった指標で判断する)。しかし、どの論文も、単純にAlphaFold2の予測構造を使えば良いわけではなさそうだという結論であった。 link.springer.com www.cell.com
実は、実験的に解いたPDBのタンパク質の立体構造でも、点数が必ずしも良いというわけではない。化合物が結合するときは、タンパク質も多かれ少なかれ形が変わっているのである。なにも結合していないタンパク質の構造を「アポ構造」と呼び、何かが結合した状態の構造を「ホロ構造」と呼ぶ*2。雰囲気だけで言えば、なにも結合していないアポ構造では結合ポケットの形が狭まってそうであり、なにかが結合したホロ構造であれば結合ポケットが多少広がってそうである。実験的に解いた立体構造がどっちなのかでバーチャルスクリーニングの点数が変わり、平均的には何かが結合したホロ構造のほうが点数が良いという報告が多い(モノによる)。 tonets.hatenablog.com
ではAlphaFoldはどうなのか。実は、AlphaFold2がデフォルトで予測した構造の多くはアポ構造に近く、ポケットが狭い状態になっていそうである(もちろんモノによる)。そのため、AlphaFold2を用いる場合は、デフォルトの予測構造だけを用いるのではなく、予測を実行するときのパラメーターをいろいろ変えてみることが提案されている。知ってる方向けに言えば、たとえばmax_msa、seed、dropoutである。さらには、AlphaFold2の処理の最初に行われる多重配列アラインメントの中身を一部壊してみるのも有効である。いずれにしても、実際にAlphaFold2の予測構造を用いてバーチャルスクリーニングを行うときは、いくつかの異なる予測構造を生成しておいて、答え合わせの点数が良さそうか悪そうかをまず試してみると良い。我々はAlphaFold2の多重配列アラインメントの壊し方を変えて、出てきた構造でドッキングし、答え合わせの点数を見てまた壊し方を変えて、ドッキングし、・・・という繰り返しを遺伝的アルゴリズム (genetic algorithm) という方法で行うことで、自動的に良い立体構造を選ぶ方法も作ってきた。 www.sciencedirect.com
なお、ここで勘の良い読者は気づいたと思うが、「答え合わせ用の活性化合物/非活性化合物が既に存在する」という状況は、創薬的にはとても恵まれている状況、あるいはもはや取り組む意味のない状況かもしれない。しかし、ノーヒントでは良い立体構造を選択することは難しい。タンパク質の直接の結合活性実験等のデータでなくても、細胞実験の結果や、形が似たタンパク質の活性化合物の情報など、なんらかのヒントをかき集め、なんとか答え合わせに近いことができる状況を準備しておくことは重要だろう。
AlphaFold3と低分子化合物の合理的創薬
AlphaFold2の存在が最初に発表された2020年11月から3年ちょっと経って、AlphaFold3が登場した。AlphaFold2はアミノ酸の塊しか扱えなかったが、AlphaFold3ではタンパク質以外の分子や金属、DNA/RNAなども入力できるようになり、さまざまな複合体(ヘテロ複合体)が予測できるようになった。 zenn.dev
ということは、先のアポ/ホロの問題は、これで解決するように思える。なんらかの化合物とタンパク質をAlphaFold3に放り投げれば、複合体構造が予測され、その構造はホロ構造になっていそうである。
その推測は概ね正しい。が、2つの疑問がある。1つ目は、予測された化合物の結合部位が本当に正しいかという問題、2つ目は、どんな化合物を入力すればよいのかという問題である。 1つ目の話はAlphaFold3の論文内でもある程度評価されているが、体感では5割くらいの正解率だと思う(有識者の皆様、どうだろうか?)。どちらかというと、AlphaFold3が「知ってる」結合部位への予測は割と正しくできることが多く、AlphaFold3が「知らない」結合部位への予測はたぶん難しいと思う。(すなわち、本来の基質結合部位などへの予測は得意そうだが、基質結合部位以外の部位へ結合して作用するような(アロステリックな)結合の予測は、すでにPDBに登録された例が無ければおそらく無理かと思われる。)また、複数の結合部位の存在をAlphaFold3が知ってる場合にも間違えることがありそうである。
2つ目の話は検証を行った結果を我々のグループで報告している。当然ながら結合する化合物の情報を入力した方が平均的なバーチャルスクリーニングの成績は良かった。結合する化合物の情報がない場合は、なにも化合物を入力しないよりは分子量400~500くらいの上市薬をテキトウに選んで突っ込んでおいた方が良さそうである。いずれにしても、ここでも「点数が算出できる状況」を想定しているので、AlphaFold2を使う場合と同じ問題は依然として存在する。 www.sciencedirect.com
Boltz-2と低分子化合物の合理的創薬
AlphaFold3が登場したことは製薬企業からの注目も大きかったが、商用利用ができないライセンスのため他のツールの利用が検討された。AlphaFold3の登場後、さまざまな似たツールが開発され発表されたが、中でもMITのグループが開発したBoltz-1は、オープンソースライセンスであり自由に活用ができるということで普及が進んだ。なにより、Boltz-1独自の機能として、化合物が結合する残基を指定して予測する機能があり、狙いたい結合部位を決めてバーチャルスクリーニングをするような状況に有用である。 https://www.biorxiv.org/content/10.1101/2024.11.19.624167v1
その後、Boltz-2という後継ツールが公開された。Boltz-1とBoltz-2は、立体構造予測という面ではほとんど性能の違いはないが、Boltz-2では構造予測のついでに化合物の親和性の予測も可能になった。すなわち、IC₅₀の予測値を出してくれるわけである。これにより、Boltz-2だけでバーチャルスクリーニングができるようになった。どのくらいの正答率かは論文で検討がされており、ドッキングシミュレーションによるバーチャルスクリーニングよりも遥かに高い正答率が報告されている(分子動力学シミュレーションによる結合自由エネルギー計算に匹敵すると言われているが、実際は結合自由エネルギー計算の方が優秀そうである)。どのくらいの質の予測ができるのかは元論文やこちらのウェブ記事を見てもらうのが良いが、最近の学会(CBI学会2025年大会など)でも製薬企業のin houseデータを当てさせてそれなりに高い正答率になったとの報告があり、AIの訓練データに含まれていないような化合物でもうまく当てられそうな期待がある。製薬企業各社とも、どんな場合にBoltz-2がうまくいくのか・いかないのか、どんなパラメーターを選べば良いのかなど、検討を進めている(Tokyo-1 Communityで実施されたTokyo-1 Drug Discovery Hackathonなどの取り組みがある)。 xeureka.co.jp
AlphaFoldとペプチド創薬
2024年ノーベル化学賞のテーマは、タンパク質立体構造予測とタンパク質デザインであった。立体構造をアミノ酸配列から予測するのが「順問題」とすれば、タンパク質デザインは逆問題、すなわち望みの構造や機能になりそうなタンパク質のアミノ酸配列を予測する問題と言える。ノーベル化学賞を受賞したDavid Bakerらが先駆的な研究を数多く進めているが、現在ではデザインフローの中にAlphaFoldなどの立体構造予測技術が取り入れられ、タンパク質デザインの成功率が格段に上がっている。
 (From sequence to function through structure: Deep learning for protein design - ScienceDirectより, CC-BY 4.0)
doi.org
(From sequence to function through structure: Deep learning for protein design - ScienceDirectより, CC-BY 4.0)
doi.org
特定のタンパク質に結合しそうなペプチドを設計するという問題でも、同じ枠組みが利用できる。AlphaFoldを使いながら良さそうなタンパク質-ペプチド複合体予測構造が得られるようなペプチド配列を探索し、良い感じのペプチドになったらOKという発想である。AlphaFold2が登場した当初は標準アミノ酸20種でかつ直鎖状、すなわち「フツウ」のペプチドしか扱えなかったが、Head-to-Tail環状ペプチドへの対応(cyclic offset法)、ラリアット型などの環状ペプチドへの対応 (HighFold)、N-メチル化やD体などの特殊アミノ酸への対応 (HighFold-MeD) などの拡張手法が提案され、また現在ではAlphaFold3やBoltz-1による特殊アミノ酸・特殊形状のペプチドの予測も研究が進んでいる。 boltz.bio
AlphaFoldと抗体創薬
抗体もアミノ酸の塊だと思えば、AlphaFoldでうまく予測したり設計したりできそうに思われる。半分正解であるが、抗原抗体複合体構造の予測精度、特に抗原を認識する相補性決定領域 (complementarity-determining region, CDR) が多様で、構造がとても柔軟であるために、構造予測が難しいという点が課題である。AlphaFold3では少しマシになっているものの、詳細な抗原抗体複合体の構造予測はまだそこまで精度が高くないため、構造予測に基づく配列設計の成否が未知数なのだ。
我々はCDRの配列に絞ってうまく配列を設計する方法を検討してきた。強化学習に基づくアミノ酸配列生成手法を用い、結合親和性の計算値ΔΔGをラベルとして学習させて抗原に特異的に結合しそうな配列を生成する枠組みを2024年に発表 (NeurIPS 2024 MLSB workshop) し、次いでタンパク質のアミノ酸配列から事前学習されたタンパク質言語モデルを用い、結合親和性の計算値ΔΔGをRoLAと呼ばれる技術でフィードバックして言語モデルをファインチューニングすることで抗原に特異的に結合しそうな配列を生成する枠組み (J Chem Inf Model 2025) を2025年に発表した。
世界的に見ても計算による抗体設計の研究は激化している。2024年にはDavid BakerらがRFantibodyと呼ばれる方法を報告しており(2025年 Nature掲載, くろたんくLabさんの解説)、ヴァンダービルト大学のIvelin Georgievらはタンパク質言語モデルに基づく生成モデルのMAGEという方法で抗体を自在に設計する技術を報告した(2025年 Cell掲載)。また、全容は不明だが、Chai Discovery社が開発するChai-2や、Latent Labs社が開発するLatent-X2といった新しいツールの開発・利用も進んでいるようである。このあたりの解説はバイオインフォマティクスVTuberの澪乃ゆいさん @mionoyui の記事(創薬 (dry) Advent Calendar 2025 3日目)が参考になるかもしれない。 zenn.dev
おわりに
AlphaFoldは構造予測の時代を変えたが、万能ではない。単体の立体構造としてはおおよそ9割ぐらいは当たっている感触があるが、創薬における肝心なところ、例えば点変異、化合物の結合、アロステリー、誘導体のSAR、抗体のCDR、非天然アミノ酸などでは、まだまだという印象もある。何より、AlphaFoldだけでどうにかなるわけではなく、AlphaFoldをもとにした新しいツールや、言語モデルなどほかのAI技術、また分子シミュレーション技術も、重層的に活用がされている。言うまでもなくwet実験はさらに重要になってきており、dry-wetのサイクル(DMTAとかDBTLとか言われるやつ)をうまく回していくことが、リード化合物創出には不可欠である。
ということで、AlphaFoldは創薬の役に立っていると信じているが、ウマく付き合っていく必要がありそうである。
*1:実験で解いた構造はProtein Data Bank (PDB) というデータベースに登録する決まりがあるので、すでに解かれた構造が知りたいときはPDBを見れば良い。
*2:アポ/ホロという言葉は、元来は酵素とその補因子に対して使われてきたものであり、補因子がない状態の酵素をアポ酵素 (apoenzyme)、補因子を含む完全な構造をホロ酵素 (holoenzyme) と呼んでいた。ChatGPT 5.2によれば、酵素・補因子のapo/holo呼称は1930年代に定着し、それ以外のタンパク質や分子へ一般化してapo/holoと呼ぶようになったのは1980年代後半〜1990年代初頭の構造生物学の論文からとのことである。
とある准教授の2024年の年収
とある助教の年収の記事は昨年で終わりました。今年からとある准教授の年収です。 tonets.hatenablog.com
2024年12月の給与明細

一部解説
- 裁量労働制という制度なので、定時や残業という概念はありません。
- いわゆる新年俸制(退職金を退職時にもらう年俸制)です。
- 基本給が年に12回払われます。
- ボーナスが6月と12月にあります。
- 子どもを2人扶養しているので扶養手当を頂いています。
- 都市手当というものが存在します。(基本給+業績給(一)(ナニモノ?)+基本給の調整額(ナニモノ??)+高校教員調整額+管理職手当+扶養手当)×18.8%で計算されます。
- 競争的研究資金からPI人件費を100万円出していますが、業績付加給付金という形で98万円支給されています。
- いわゆる住宅ローン控除をほぼフルで活用しています。
- 一般生命保険料控除だけやってます。(iDeCoやってません)
前年度比で+66万円でした。 評価で変動する分は、今年の1月に准教授になったのもあって多分評価対象外のため、よくわかりません。
とある助教の2023年の年収
1年ぶりになってしまったブログ記事です。
2023年12月の給与明細
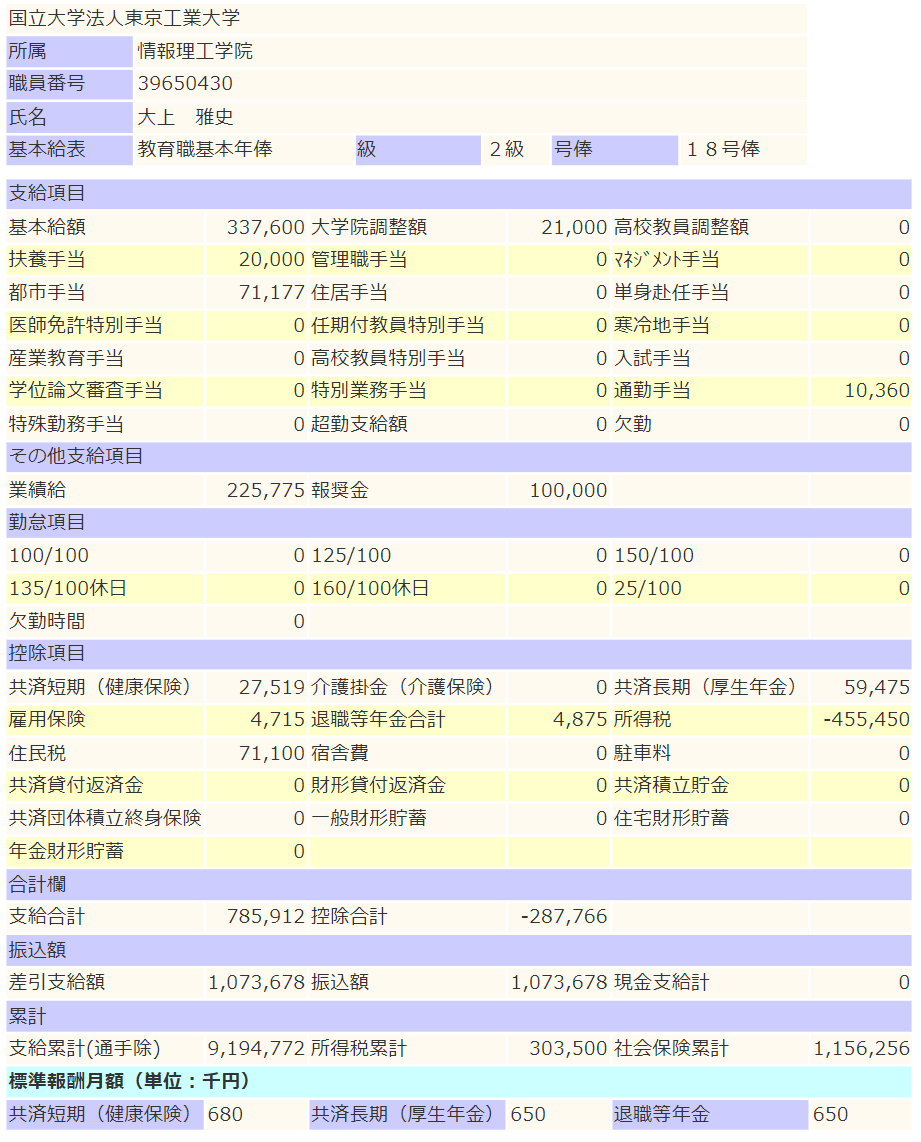
一部解説(12月分に記載のないものもあります)
- 裁量労働制という制度なので、定時や残業という概念はありません。
- いわゆる旧年俸制(退職金を先に払ってる年俸制)です。
- 基本給が年に12回払われます。
- ボーナスという概念が月々支払われる業績給に吸収されています。業績給は均されて定額が年に12回支払われます。
- 業績給に退職金を含んでいます。(通常の課税対象です)(退職時にもらえる退職金は0です)
- 任期付の教員ですが任期付教員特別手当が出ません。
- 子どもを2人扶養しているので扶養手当を頂いています。
- 都市手当というものが存在します。(基本給+大学院調整額+高校教員調整額+管理職手当+扶養手当)×18.8%で計算されます。
- 競争的研究資金からPI人件費を100万円出していますが、業績付加給付金という形で98万円支給されています。
- 東工大教育賞という賞をもらったときの副賞として報奨金を10万円もらえました。(もちろん課税対象です)
- いわゆる住宅ローン控除をほぼフルで活用しています。
- 一般生命保険料控除だけやってます。(iDeCoやってません)
前年度比で+16万円ですが、報奨金を引いたら賞味+6万円でした。
(とある助教の年収の記事は、今回で一旦終了としたいと思います。)
2024.1.1追記
2024年1月1日付で、東京工業大学 情報理工学院 助教から准教授に昇任しました。テニュアトラックの審査をクリアした形です。任期が無くなること以外には全く何も変わりませんが、引き続き皆様よろしくお願い致します。
— Ohue M/大上雅史 (@tonets) 2023年12月31日
とある助教の2022年の年収
35歳助教氏ワイ、年収900に副業と印税あるので待遇そのものは相変わらずあんまり困ってないが、任期付きなのと、退職手当は無い。 https://t.co/7x819rnBrc
— Ohue M/大上雅史 (@tonets) 2022年12月26日
こちらのエビデンスです。
(2021年のものはこちら)
tonets.hatenablog.com
2022年12月の給与明細

ペプチドとドッキングとAlphaFold
この記事は?→創薬 (dry) Advent Calendar 2022の5日目の記事です。 adventar.org
10年以上の年月と3,000億円超の費用を要する医薬品開発の期間とコストを削減するため、創薬の様々な場面でコンピュータによるインシリコ(in silico)解析が導入されている。特に通常の低分子化合物では、バーチャルスクリーニングによるヒット化合物の発見、標的分子と活性リガンドの構造情報を利用した分子設計によるリード化合物の最適化、物性や動態の予測・最適化に至るまで、様々な課題に対して計算論的手法が開発されてきた。かたやペプチドについては、低分子と比較するとやや「出遅れていた」状況であった。だが、近年の技術革新によりインフォマティクス技術を基盤としたペプチドの設計が実現しつつある。以下ではペプチド創薬に活用できる、ペプチドを対象としたインシリコ(in silico)解析技術を紹介する。
タンパク質-ペプチドドッキング
ドッキングシミュレーション(単にドッキングとも言う)は、標的となる分子と目的の分子の複合体構造を推定し、併せて標的親和性を計算によって評価するための方法論である。標的としてはタンパク質やRNA、目的の分子としては低分子や金属イオン、糖鎖などが該当し、たとえばタンパク質と低分子(タンパク質-リガンドドッキング)、タンパク質とタンパク質(タンパク質ドッキング)、タンパク質と金属イオン、タンパク質と糖鎖など、個々に手法が開発されている。このうち、タンパク質とペプチド(主に数残基~20残基程度のサイズ)を対象とした技術をタンパク質-ペプチドドッキングと呼ぶ。
タンパク質-ペプチドドッキングは、標的タンパク質に対するペプチド分子の結合様式(複合体構造)を推定し、その結合親和性をエネルギースコア等の値として評価するような手法を言う。いくつかのソフトウェアが提案されているが、なかでもAutoDock CrankPep*1が、数残基~20残基くらいまでのペプチドに対して精度良く複合体構造(図1)を推定可能であるとされている。

ドッキングソフトウェアから計算されるエネルギースコアの値の良し悪しで、ペプチドの標的タンパク質に対する結合能が評価できる。エネルギースコアは「タンパク質のどこにどんな形で結合しそうか」を評価するための計算値であるが、たとえば「目的のペプチドが、他のすでに知られている結合ペプチドや、ランダムな配列のペプチドと比較して、どのくらい結合能が高い(or低い)のか」を推定すること、すなわちバーチャルスクリーニングにも活用できる。
なお、ほとんどのドッキングソフトウェアでは直鎖かつ標準アミノ酸20種で構成されたペプチドしか扱うことができなかったが、最近ではD体アミノ酸や修飾のあるアミノ酸などの非標準アミノ酸を含んだペプチドや、ペプチド鎖の末端や途中で環状型になっているペプチド(環状ペプチド)についても入力として扱えるソフトウェアが増えてきた。AutoDock CrankPep(環状ペプチドに対する評価は文献*2を参照)やHADDOCK*3は、環状ペプチドに対するドッキングシミュレーションを実行可能なソフトウェアとしても使われている。
ところで、タンパク質立体構造予測の分野ではAlphaFold2による予測精度の革新があったことは記憶に新しい。実は現在のAlphaFold2は、タンパク質複合体(ホモ/ヘテロオリゴマー)の構造予測が可能となっており、タンパク質ペプチド複合体構造の予測にも使えそう(図2)だということが複数のグループによって検証されてきた*4*5*6。

世界で初めてAlphaFold2によるペプチドドッキングを報告したツイート。残念ながら後の論文からはreferされず。(英語でtweetしておけばよかった)あ、AlphaFold2でペプチドドッキングできちゃった pic.twitter.com/BkNs6davJR
— Ohue M/大上雅史 (@tonets) 2021年7月20日
特にこれらの報告の中で、従来のドッキングと同様にペプチドのバーチャルスクリーニングができる可能性が示唆されている。ただし、AlphaFold2の制限として直鎖・標準アミノ酸のペプチドしか扱うことはできないことに注意が必要である。さらなる応用として、たとえば2つのペプチド配列を入力して、より結合が強い方がタンパク質に結合するような結果が得られるという、競合ドッキング法(competitive binding)と呼ばれる方法も提案されている*7(図3)。
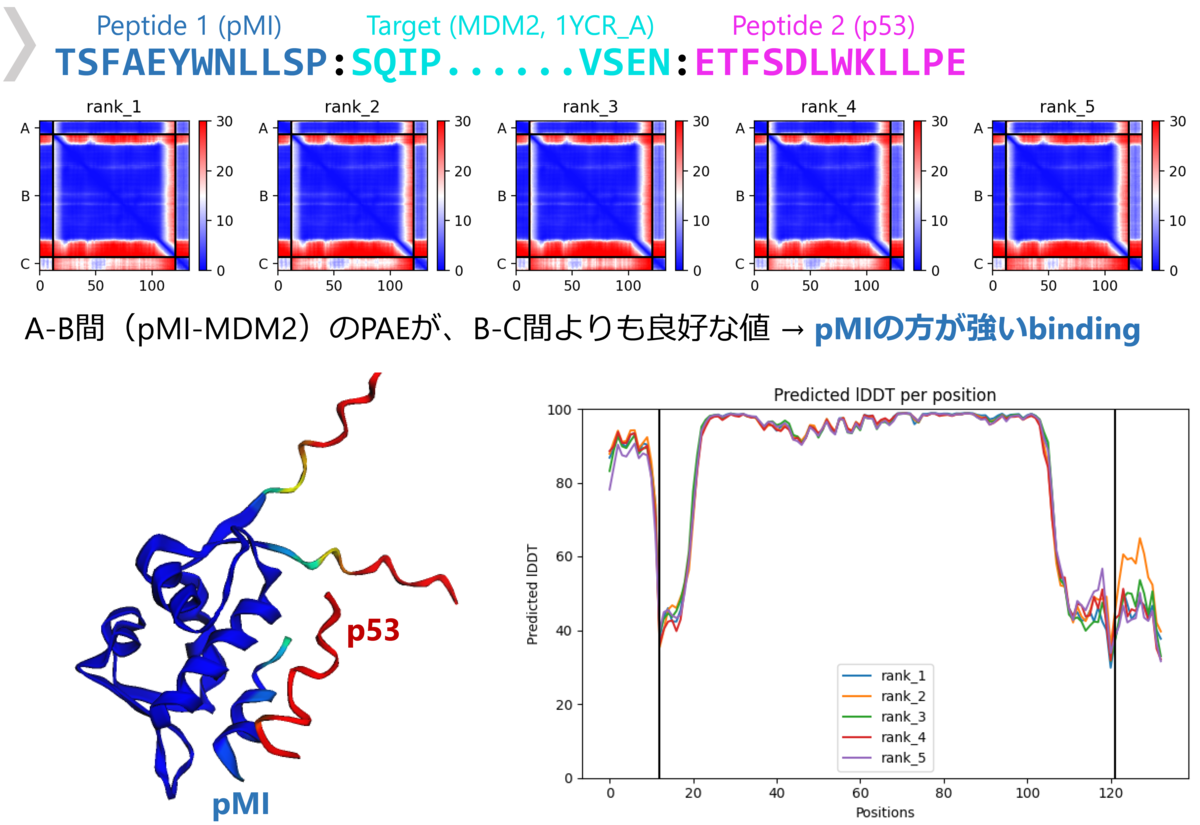
AlphaFold2によるペプチドデザイン
計算によって標的タンパク質に対するペプチドの結合能を評価することができるということは、手元にあるペプチド配列の比較だけでなく、あらゆるペプチド配列をソフトウェアに入力していくことで、標的タンパク質に対して結合能が高いペプチド配列が見つけられるということになる。ただしこのような発想で実際に網羅的にペプチド配列のスクリーニングを行う場合には、ペプチド残基長nに対して20ⁿ回の計算をする必要がある。10残基のペプチドだけを考えたとしても20¹⁰ ≒ 10兆通りのペプチド配列の評価が必要になり、これは非現実的である。対して、ランダムな配列からスタートし、予測の結果と欲しい構造との差を勾配としてフィードバックにして、フィードバックの結果から次の配列を選んでいくという方法(Hallucination法)*8が提案されており、予測値が良くなる方向にバイアスをかけて配列を選択することで、全通りの計算を避けて妥当なペプチド配列を生成することが可能になっている。
このアイデアに基づいて、Hallucination論文やRoseTTAFoldの著者でもあるSergey Ovchinnikovらによって実装されたAfDesignのbinder hallucination法で、実際にペプチド配列が生成できるようになっている。AlphaFold2の出力するペプチド複合体予測の評価値(pLDDTやPAEなど)を良くする方向にペプチド配列をサンプリングしていくことで、AlphaFold2的に良いと考えるペプチドを現実的な計算時間で生成するという仕組みである。実際にAfDesignを実行すると、実際に図4のようにペプチド配列が決定されていく様子を動画で出力することができる。

Adding support for binder hallucination if anyone wants to try! (Code is very experimental, not intended for practical use... only use for art/science) 😀https://t.co/OOPp8kSu2Z pic.twitter.com/WUu9LGKIwp
— Sergey Ovchinnikov @ NeurIPS 🇺🇦 (@sokrypton) 2022年2月3日
AlphaFold2による水溶性ペプチドデザイン
ところが、AfDesignによって生成された配列を確認すると、難水溶性のペプチドが多くを占めていた。タンパク質間相互作用の相互作用面は一般に疎水領域であり、AlphaFold2 (AlphaFold-Multimer) も相互作用面に共起しやすい残基の関係を学習していると考えられることから、タンパク質の表面に結合するペプチド配列をAlphaFold2によって設計しようとすると疎水領域を構成するように、すなわち疎水性残基を多用したペプチド配列が選ばれやすくなっているのだと解釈できる。だが、後の生化学実験などを考える上ではペプチドの水溶性は重要である。
我々はこの問題を解決するために、AlphaFold2の評価値に加えてhydropathy indexなどのアミノ酸に関する物性評価指標を導入し、「疎水性アミノ酸はあまり使わないように」といった形で残基の使用頻度を制御することで、適切なペプチド配列を生成する手法「Solubility-AfDesign」を提案した*9。Solubility-AfDesignによって、実際に標的結合能を維持したまま水溶性を向上させるペプチド配列の予測に成功し(図5)、具体的な複合体構造モデルとともに提示することができるようになった。この論文は日本語で以下に解説があるので、興味を持った方は読んで頂ければ幸いである。 blacktanktop.hatenablog.com
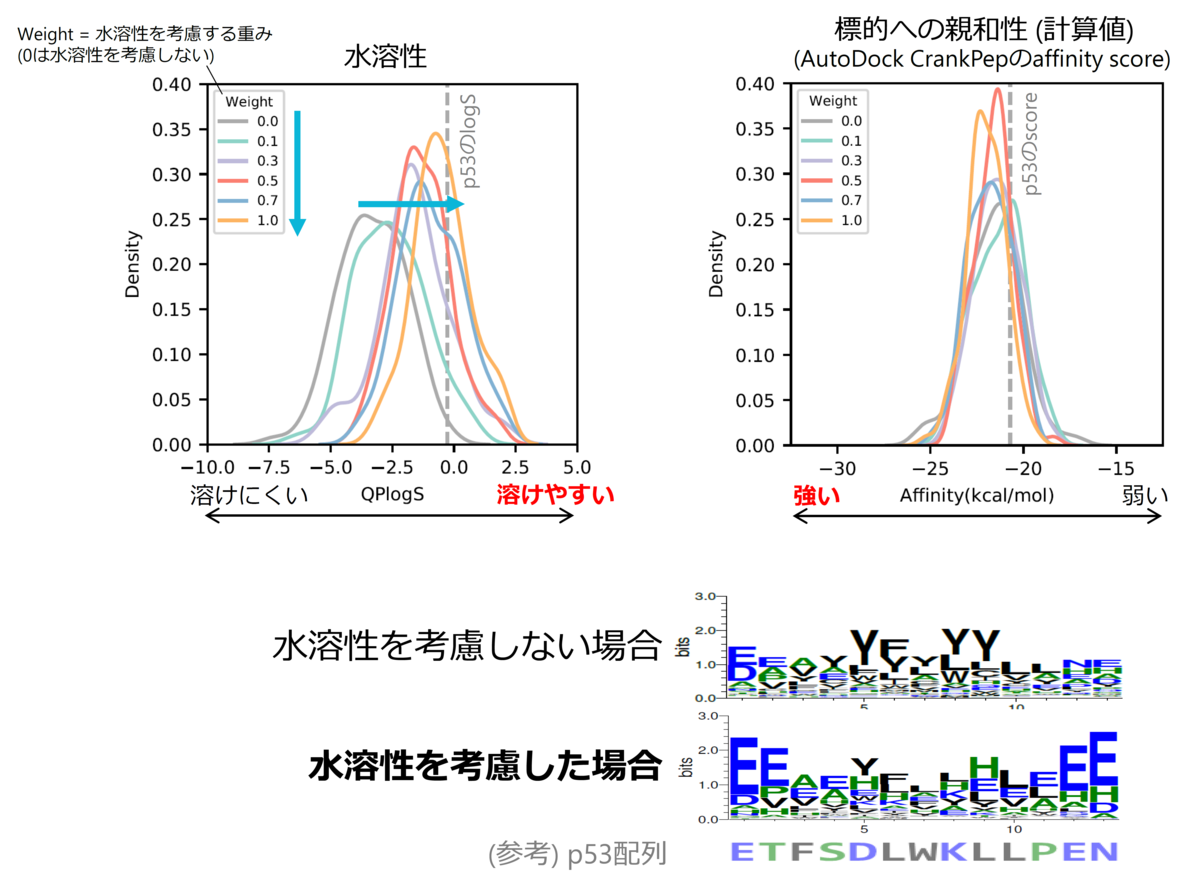
まとめ
本稿ではペプチドに関するin silico技術を紹介したが、ペプチドの計算技術・設計技術はまだまだ発展を続けている段階である。タンパク質構造、低分子、ペプチドと、それぞれでAIやシミュレーション技術が展開されてはいるが、まだまだin silicoが追いついていない領域でもあるかと思う。AlphaFold2やChromaのように突然すごいAI技術が降ってくるかもしれない。どれがイケててどれは微妙なのか、目利きも重要になるかと思う。今後の発展に目が離せない。
Today we introduced Chroma, a generative model that creates new proteins & protein complexes given geometric & functional constraints. It learns to transform unstructured, random 3D shapes into #protein molecules, which can have tens of thousands of atoms. https://t.co/cORRnRKnfB pic.twitter.com/2OUk9AOQuj
— Generate Biomedicines (@generate_biomed) 2022年12月1日
*1:Zhang Y, Sanner MF. AutoDock CrankPep: combining folding and docking to predict protein-peptide complexes. Bioinformatics. 2019;35(24):5121-5127. doi: 10.1093/bioinformatics/btz459.
*2:Zhang Y, Sanner MF. Docking Flexible Cyclic Peptides with AutoDock CrankPep. J Chem Theory Comput. 2019;15(10):5161-5168.
*3:Charitou V, van Keulen SC, Bonvin AMJJ. Cyclization and Docking Protocol for Cyclic Peptide-Protein Modeling Using HADDOCK2.4. J Chem Theory Comput. 2022; 18(6):4027-4040.
*4:Ko J, Lee J. Can AlphaFold2 Predict Protein-Peptide Complex Structures Accurately? bioRxiv 2021.07.27.453972, 2021.
*5:Tsaban T, Varga JK, Avraham O, Ben-Aharon Z, Khramushin A, Schueler-Furman O. Harnessing protein folding neural networks for peptide-protein docking. Nat Commun. 13(1):176, 2022.
*6:Johansson-Åkhe I, Wallner B. Improving peptide-protein docking with AlphaFold-Multimer using forced sampling. Front Bioinform, 2, 959160, 2022.
*7:Chang L, Perez A. AlphaFold encodes the principles to identify high affinity peptide binders, bioRxiv 2022.03.18.484931, 2022.
*8:Anishchenko I, Pellock SJ, Chidyausiku TM, Ramelot TA, Ovchinnikov S, Hao J, Bafna K, Norn C, Kang A, Bera AK, DiMaio F, Carter L, Chow CM, Montelione GT, Baker D. De novo protein design by deep network hallucination. Nature. 2021; 600(7889):547-552.
*9:Kosugi T, Ohue M. Solubility-aware protein binding peptide design using AlphaFold. Biomedicines, 10(7): 1626, 2022.
タンパク質間相互作用 (PPI) を標的とする化合物の設計
この記事は?→創薬 (dry) Advent Calendar 2022の4日目の記事です。 adventar.org
タンパク質間相互作用 (protein-protein interaction, PPI) が創薬標的として注目されるようになって久しい。一般に難しい標的とされているPPIであるが、近年は様々な計算技術とともにPPI標的化合物をうまく設計するための方法が発展してきている。この記事では、最近のPPI標的化合物設計におけるin silico手法を紹介したいと思う。
タンパク質間相互作用(Protein-Protein Interaction)を狙う低分子化合物
タンパク質間相互作用 (protein-protein interaction, PPI) はあらゆる生命現象に介在しており、創薬標的として高い注目を集めている。ヒトにおけるPPIは現時点で883,507件報告*1されており、今なお新たなPPIの発見が報告されている。実際にはこれらのPPIが全て潜在的な創薬標的となるわけではなく、当然ながら様々な理由により創薬標的として適さない(‘undruggable’な)PPIも多いため、実際に創薬標的とされているPPIの種類はわずかである。しかしながら、例えば複合体の立体構造が解かれてラショナルドラッグデザインが可能となっていく、低分子では阻害が難しかったPPIでも近年の新規医薬モダリティの発展によって阻害できるようになっていく、といった、近年および今後の状況の変化により潜在的なdruggable PPIはますます増えていくことが想定され、多様なメカニズムを持つPPIは今後ますます創薬研究に影響を及ぼすことと思われる*2。
従来の分子標的薬設計においては、タンパク質構造上の特定の結合サイト(例:基質結合ポケットなど)に対して化合物構造を設計するが、PPIを標的とする場合はより広い表面領域(相互作用インターフェース)を狙う必要がある。図1にPPIの例としてInterleukin-2とInterleukin-2受容体αの複合体構造を示す。

タンパク質間相互作用を標的とする化合物の設計指標
経口医薬品に関する設計指標 – RO5 と QED
LipinskiのRule-of-Five (RO5)*4は、経口医薬品が満たすべき性質の経験則としてよく知られている。RO5は以下の4つの記述子によるルールで構成されている。
いずれのルールも数字の5または5の倍数にちなむことから “Rule-of-Five” と名付けられている。このルールを2つ以上満たさないような化合物は吸収が悪く、最終的に医薬品になりづらいとされている。実用上は、LogP値は実験値ではなく計算による推定値を用いることがほとんどであり、CLogPやMolLogPがよく用いられる。
RO5は選ばれている分子記述子の計算が容易であり、記述子の種類も様々な物性との相関がよく知られているものであるため、創薬化学者にとっても理解しやすく、受け入れられやすい指標である。一方で、ルールに合うか合わないかという2値での分類をしているため、たとえば分子量501の分子と分子量1000の分子は同様に除外されてしまうという問題がある。実際には分子量501で若干オーバーしていたとしても全く医薬品として望ましくないというわけではないため、適合度合いをより定量的に評価することが重要である。このような医薬品らしさを定量評価する指標として、Bickertonらが2012年に提唱したQED*5がある。QEDでは771個のFDA承認薬(経口医薬品)から次の8種類の記述子の分布を求め、分布の頂点の値を取るような記述子の値を持っている分子は「薬らしい」と定義して0から1の値として定量化をしたものである。
- 分子量 (MW)
- オクタノール/水分配係数(脂溶性) (LogP)
- 水素結合ドナーの数 (HBD)
- 水素結合アクセプターの数 (HBA)
- 極性表面積 (PSA)
- 回転可能結合数 (ROTB)
- 芳香環の数 (AROM)
- 忌避構造の数 (ALERTS)
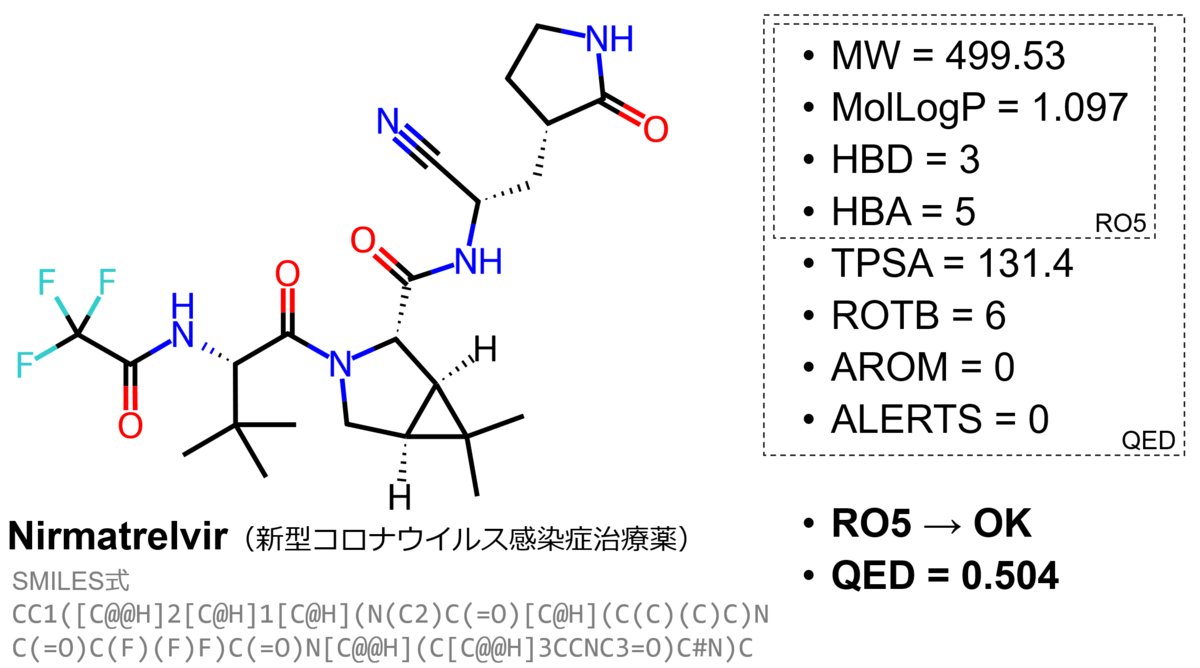
QEDに使われている記述子を見るとわかる通り、QEDで用いる記述子はRO5と共通する。これらの記述子も簡単に計算できるため、QEDは分子設計の上でよく用いられている指標となっている。たとえば新型コロナウイルス感染症治療薬として使われている合剤パキロビッド®パックに含まれるプロテアーゼ阻害剤ニルマトレルビル (Nirmatrelvir) について、各記述子およびRO5とQEDを計算したものが図2である。QED = 0.504という値はRO5を全て満たしている医薬品のQED値の分布から見るとやや低いものの、分布から外れた値というわけではない(詳細はQED論文のFigure 2(c)を参照)。
なお、それぞれの記述子の計算にはPythonライブラリであるRDKit version 2022.9.1を用い、以下のコードで計算した。特にLogPは推定値であるMolLogP、PSAは推定値であるトポロジカルPSA (TPSA) で代用した。
from rdkit import Chem m = Chem.MolFromSmiles(r'CCOC(=O)C1=C(N(C2=CC(=C(C(=C21)CN(C)C)O)Br)C)CSC3=CC=CC=C3') Chem.Descriptors.MolWt(m) # MW Chem.Descriptors.MolLogP(m) # LogP Chem.Descriptors.NumHAcceptors(m) # HBA Chem.Descriptors.NumHDonors(m) # HBD Chem.Descriptors.TPSA(m) # PSA Chem.Descriptors.NumRotatableBonds(m) # RotB Chem.Descriptors.NumAromaticRings(m) # AROM Chem.QED.properties(m).ALERTS # ALERTS # Chem.QED.properties(m) # これで全部出せる。ただしHBA/HBDの値はNumHAcceptors/NumHDonorsの値と異なる場合がある。(!?)
PPI標的化合物に関する設計指標 – RO4とQEPPI
RO5およびQEDは、経口医薬品の持つ統計分布や経験則を用いた指標であった。一方で、PPI標的化合物には通常の経口医薬品と比べても「分子量が大きめ」といった異なる特徴が求められる。実際に、MorelliらはPPI阻害剤39個のデータからPPI阻害剤が持つ記述子の経験則をRule-of-Four (RO4) という形で以下のようにまとめた*6。
いずれのルールも4にちなんでいることから “Rule-of-Four” と呼ばれている。RO4を満たすような化合物はPPI阻害剤になりやすい性質を有していると考えられる*7。
一方で、RO5とQEDの関係と同様に、定量的に判断できる指標は重要である。我々はQEDの考え方を参考にして、記述子の統計値をもとに「PPI標的化合物らしさ」を定量化できる指標としてQEPPI (https://github.com/ohuelab/QEPPI) を開発した*8。QEPPIでは、関数モデリングに先立ち、Morelliらが用いていたデータよりもより広範なデータを収集するため、論文等からPPI標的化合物を登録しているiPPI-DBを用い、ここから冗長性を省いた1,007化合物を選択した。これらのデータについてQED記述子のうちALERTSを除く7つの記述子の分布*9を求め、QEDと同様に関数モデリングを行って0から1の値としてPPI標的化合物らしさを定量化した。図3にiPPI-DB化合物に対するそれぞれの記述子のヒストグラムを描画し、分布関数を重ねたものを示す。QEDと比較して分布の山がいずれの記述子でも値が大きい方にずれていることがわかるかと思う。
QEPPIの具体的な例として、たとえばコロナウイルスのスパイクタンパク質とヒト細胞表面のACE2タンパク質との相互作用を阻害することが知られている抗ウイルス薬ウミフェノビル (Umifenovir) は、QED = 0.376と低めのQED値を示す一方で、QEPPI = 0.869と高いQEPPI値を示す(図4)。また、実際に開発されている臨床試験段階のPPI標的化合物についてQEPPI値を計算したところ、比較的高い値を示すこともわかった(図5)。
なお、それぞれの記述子の計算にはRDKit version 2022.9.1を用い、QEPPIは以下のPythonコードを用いて計算した。
from rdkit import Chem import QEPPI as ppi m = Chem.MolFromSmiles(r'CCOC(=O)C1=C(N(C2=CC(=C(C(=C21)CN(C)C)O)Br)C)CSC3=CC=CC=C3') q = ppi.QEPPI_Calculator() q.read() q.qeppi(m) # QEPPI #Chem.rdMolDescriptors.CalcNumRings(m) # * RING の計算は CalcNumRings()



タンパク質間相互作用向け化合物ライブラリー
PPIライブラリー
PPI標的薬の設計には、標準的な化合物ライブラリーではなく、PPI標的薬になりそうな化合物を集めたフォーカストライブラリーであるPPIライブラリーを用いる方が、ヒット化合物を得られる可能性が高くなると考えられる。単純に化合物ライブラリーからRO4を満たす化合物を選んで使うだけでも簡易的なPPIライブラリーとして活用できるが、特にタンパク質の相互作用面の特徴などから提案されたPPIライブラリーが、化合物サプライヤーから提供されている。以下に代表的なPPIライブラリーを4つ紹介する。
Enamine社が提供するPPIライブラリー (https://enamine.net/compound-libraries/targeted-libraries/ppi-library) はその代表例である。Enamine社のPPIライブラリーは、複数のタンパク質複合体構造を解析し、PPIに特徴的な二次構造やモチーフに特化して設計された化合物を集めたものであり、さらに忌避構造を含まないようにフィルタリングされ、実際に合成サンプルとして得られている化合物を約4万件収載している。
ChemDiv社が提供するPPIライブラリー (https://www.chemdiv.com/catalog/focused-and-targeted-libraries/protein-protein-interaction-library/) は、ペプチドミメティクスによる化合物や大環状型化合物、スピロ化合物などの特殊な母核構造も含むライブラリーであり、約21万件の化合物からなる。
UkrOrgSyntez社が提供するPPIライブラリー (https://uorsy.com/fragments-and-targeted-libraries/uorsy-ppi-modulators/) では、RO4を満たす化合物であり、PPI相互作用面におけるホットスポット残基であるTyr/Trp/Arg残基との相互作用が期待できる官能基を含む母核が含まれた、約6,500件の化合物からなる。
Life Chemicals社は、複数の方法で構築した多様なPPIライブラリーを提供する (https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/ppi-libraries/)。実際にいくつかのPPIの相互作用面に対してドッキングシミュレーションを行うことによって構築したライブラリーや、RO4によるフィルタリングを行ったもの、公開されているPPI阻害アッセイで阻害能が確認された化合物と類似する化合物を集めたもの、独自に構築した機械学習による分類器で弁別したPPI標的化合物を集めたものなどが含まれている。
以上は化合物サプライヤーが提供するものであるが、公共データベースと論文情報から収集された既知のPPI阻害剤を含むPPIライブラリーとして、慶應義塾大学とペプチドリーム株式会社が共同で開発しているDLiP (https://skb-insilico.com/dlip) がある。DLiPでは、約15,000件のPPI阻害が期待できる化合物と、約26,000件の既知PPI阻害化合物の情報が収載されており、ウェブインターフェース上で検索や詳細閲覧が可能となっている。
PPIバーチャルライブラリー
低分子化合物の既存のライブラリーでも1億件以上の化合物が収載されている(例:ZINC15 in-stock library)が、化合物空間の多様性を考えれば化合物は無数に存在する*11。もちろんそのような化合物の中には合成が困難であったり創薬に適さない化合物も多々含まれるが、検討に値する化合物も大量に存在すると考えられる。そのような、これまでに合成されていないような化合物を計算によって「バーチャルに」生成することをde novo分子設計と呼び、機械学習の発展によって様々なde novo分子設計法が提案され、バーチャル化合物が生成されてきた。
de novo分子設計法の1つであるREINVENT*12は、リカレントニューラルネットワーク (RNN) に基づくSMILES文字列の生成モデルである。事前にChEMBLデータベースの化合物で学習したSMILES生成のための事前モデルから、あらかじめ決められた評価値が良くなる方向に学習を進める強化学習ステップによって、より良い評価値を持つ仮想的な化合物を生成するしくみとなっている。図6に我々がREINVENTを使ってQED、RO4(1ルールを満たすごとに+0.25として設計)、QEPPIの各値を評価値として与えて強化学習を行い、分子生成を行った結果を示す。QEDを良くするように設計されたバーチャル化合物は、当然ながら比較的分子量が小さく、LogPも低い傾向にある。一方でRO4を満たすように設計されたバーチャル化合物は、RO4が下限のみを定めるルールであることから分子量およびLogPが際限なく大きくなってしまうという問題がある。QEPPIでは分布から望ましい記述子の値を取るように設計されるため、QEDよりもやや分子量・LogPが大きい分子が生成されるが、たとえば分子量が1000を超えるような分子は生成されないことがわかる。我々はこのQEPPIに基づいてREINVENTで生成した化合物群を集めて、PPIバーチャルライブラリーとして公開する予定である。

まとめ
この記事では、タンパク質間相互作用を標的とする低分子設計を目指したいくつかの計算手法について紹介した。低分子化合物の創薬に役立つdry手法は、AI技術の発展とともに様々な面で発展し続けており、この記事で紹介した技術はほんの一端でしかない。タンパク質間相互作用はもはや狙えない標的ではないはずなので、これからも多くの薬が誕生することを願ってやまない。
謝辞
本記事の内容について、叢雲くすり氏 (twitter id @souyakuchan) より助言を頂きました。ここに感謝致します。
*1:BioGRIDデータベースから、生物種: Homo sapiens、Experiment type: PHYSICAL、Non-Redundant Interactions(異なる実験手法に基づく同一PPIの確認を単一カウントとする)によって検索した件数。(BioGRID version 4.4.215, 2022年11月現在)
*2:Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov, 15(8), 533-550, 2016.
*3:Thanos CD, DeLano WL, Wells JA. Hot-spot mimicry of a cytokine receptor by a small molecule. Proc Natl Acad Sci U S A, 103(42), 15422-15427, 2006.
*4:Lipinski CA. Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev, 23(1-3), 3-25, 1997.
*5:Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem, 4(2), 90–98, 2012.
*6:Morelli X, Bourgeas R, Roche P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I). Curr Opin Chem Biol, 15, 475–481, 2011. なおこの論文で言及されている2P2Iデータベースは、今はアクセスができない(汗)。
*7:Morelliらが解析したPPI阻害剤は論文等で報告されたものに基づいており、一部を除いて臨床試験段階には到達していない。その意味で、RO4が医薬品としての適正を担保するわけではないため注意が必要である(たとえば生物学的利用能は低くなることが予想される)。
*8:Kosugi T, Ohue M. Quantitative Estimate Index for Early-Stage Screening of Compounds Targeting Protein-Protein Interactions. Int J Mol Sci, 22(20), 10925, 2021.
*9:一般に分子量が大きくなるとその分忌避構造としてカウントされる部分構造を保有する確率も上がるため、ALERTSは分子量と相関し、PPI標的化合物では平均的に高い値になると考えられる。しかし、iPPI-DBに収録されている化合物のALERTSの平均値は、経口医薬品のALERTSの平均値よりも低くなるという、直感に反する結果が得られた。このことから、(初期の段階ではALERTSが低い化合物が優先的にアッセイにかけられている可能性など、)iPPI-DBのデータにはALERTSに関する選択的バイアスがあると考え、QEPPIのモデリングからALERTSを除外した。
*10:Truong, J.; George, A.; Holien, J.K. Analysis of physicochemical properties of protein-protein interaction modulators suggests stronger alignment with the “Rule-of-Five”. RSC Med Chem, 12, 1731-1749, 2021.
*11:RO5を満たす薬理活性化合物だけを考えた場合でも、潜在的に個超の化合物が存在すると推定されている。
*12:Blaschke T, Arús-Pous J, Chen H, Margreitter C, Tyrchan C, Engkvist O, Papadopoulos K, Patronov A. REINVENT 2.0: An AI Tool for De Novo Drug Design. J Chem Inf Model, 60(12), 5918-5922, 2020.