タンパク質間相互作用 (PPI) を標的とする化合物の設計
この記事は?→創薬 (dry) Advent Calendar 2022の4日目の記事です。 adventar.org
タンパク質間相互作用 (protein-protein interaction, PPI) が創薬標的として注目されるようになって久しい。一般に難しい標的とされているPPIであるが、近年は様々な計算技術とともにPPI標的化合物をうまく設計するための方法が発展してきている。この記事では、最近のPPI標的化合物設計におけるin silico手法を紹介したいと思う。
タンパク質間相互作用(Protein-Protein Interaction)を狙う低分子化合物
タンパク質間相互作用 (protein-protein interaction, PPI) はあらゆる生命現象に介在しており、創薬標的として高い注目を集めている。ヒトにおけるPPIは現時点で883,507件報告*1されており、今なお新たなPPIの発見が報告されている。実際にはこれらのPPIが全て潜在的な創薬標的となるわけではなく、当然ながら様々な理由により創薬標的として適さない(‘undruggable’な)PPIも多いため、実際に創薬標的とされているPPIの種類はわずかである。しかしながら、例えば複合体の立体構造が解かれてラショナルドラッグデザインが可能となっていく、低分子では阻害が難しかったPPIでも近年の新規医薬モダリティの発展によって阻害できるようになっていく、といった、近年および今後の状況の変化により潜在的なdruggable PPIはますます増えていくことが想定され、多様なメカニズムを持つPPIは今後ますます創薬研究に影響を及ぼすことと思われる*2。
従来の分子標的薬設計においては、タンパク質構造上の特定の結合サイト(例:基質結合ポケットなど)に対して化合物構造を設計するが、PPIを標的とする場合はより広い表面領域(相互作用インターフェース)を狙う必要がある。図1にPPIの例としてInterleukin-2とInterleukin-2受容体αの複合体構造を示す。

タンパク質間相互作用を標的とする化合物の設計指標
経口医薬品に関する設計指標 – RO5 と QED
LipinskiのRule-of-Five (RO5)*4は、経口医薬品が満たすべき性質の経験則としてよく知られている。RO5は以下の4つの記述子によるルールで構成されている。
いずれのルールも数字の5または5の倍数にちなむことから “Rule-of-Five” と名付けられている。このルールを2つ以上満たさないような化合物は吸収が悪く、最終的に医薬品になりづらいとされている。実用上は、LogP値は実験値ではなく計算による推定値を用いることがほとんどであり、CLogPやMolLogPがよく用いられる。
RO5は選ばれている分子記述子の計算が容易であり、記述子の種類も様々な物性との相関がよく知られているものであるため、創薬化学者にとっても理解しやすく、受け入れられやすい指標である。一方で、ルールに合うか合わないかという2値での分類をしているため、たとえば分子量501の分子と分子量1000の分子は同様に除外されてしまうという問題がある。実際には分子量501で若干オーバーしていたとしても全く医薬品として望ましくないというわけではないため、適合度合いをより定量的に評価することが重要である。このような医薬品らしさを定量評価する指標として、Bickertonらが2012年に提唱したQED*5がある。QEDでは771個のFDA承認薬(経口医薬品)から次の8種類の記述子の分布を求め、分布の頂点の値を取るような記述子の値を持っている分子は「薬らしい」と定義して0から1の値として定量化をしたものである。
- 分子量 (MW)
- オクタノール/水分配係数(脂溶性) (LogP)
- 水素結合ドナーの数 (HBD)
- 水素結合アクセプターの数 (HBA)
- 極性表面積 (PSA)
- 回転可能結合数 (ROTB)
- 芳香環の数 (AROM)
- 忌避構造の数 (ALERTS)
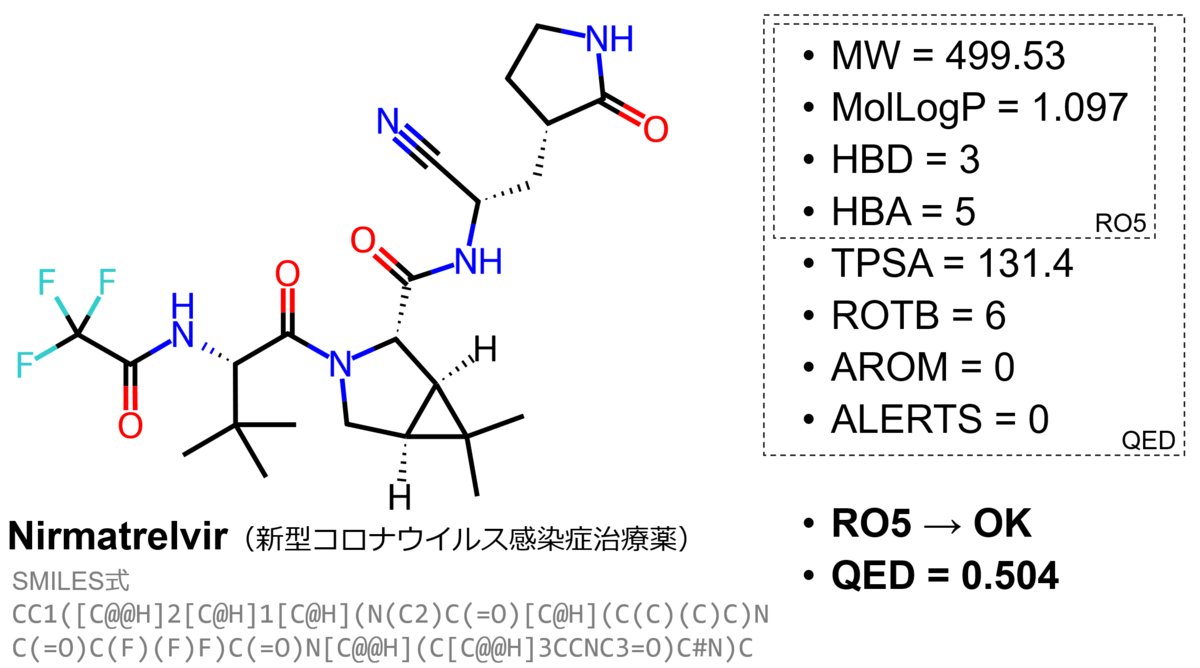
QEDに使われている記述子を見るとわかる通り、QEDで用いる記述子はRO5と共通する。これらの記述子も簡単に計算できるため、QEDは分子設計の上でよく用いられている指標となっている。たとえば新型コロナウイルス感染症治療薬として使われている合剤パキロビッド®パックに含まれるプロテアーゼ阻害剤ニルマトレルビル (Nirmatrelvir) について、各記述子およびRO5とQEDを計算したものが図2である。QED = 0.504という値はRO5を全て満たしている医薬品のQED値の分布から見るとやや低いものの、分布から外れた値というわけではない(詳細はQED論文のFigure 2(c)を参照)。
なお、それぞれの記述子の計算にはPythonライブラリであるRDKit version 2022.9.1を用い、以下のコードで計算した。特にLogPは推定値であるMolLogP、PSAは推定値であるトポロジカルPSA (TPSA) で代用した。
from rdkit import Chem m = Chem.MolFromSmiles(r'CCOC(=O)C1=C(N(C2=CC(=C(C(=C21)CN(C)C)O)Br)C)CSC3=CC=CC=C3') Chem.Descriptors.MolWt(m) # MW Chem.Descriptors.MolLogP(m) # LogP Chem.Descriptors.NumHAcceptors(m) # HBA Chem.Descriptors.NumHDonors(m) # HBD Chem.Descriptors.TPSA(m) # PSA Chem.Descriptors.NumRotatableBonds(m) # RotB Chem.Descriptors.NumAromaticRings(m) # AROM Chem.QED.properties(m).ALERTS # ALERTS # Chem.QED.properties(m) # これで全部出せる。ただしHBA/HBDの値はNumHAcceptors/NumHDonorsの値と異なる場合がある。(!?)
PPI標的化合物に関する設計指標 – RO4とQEPPI
RO5およびQEDは、経口医薬品の持つ統計分布や経験則を用いた指標であった。一方で、PPI標的化合物には通常の経口医薬品と比べても「分子量が大きめ」といった異なる特徴が求められる。実際に、MorelliらはPPI阻害剤39個のデータからPPI阻害剤が持つ記述子の経験則をRule-of-Four (RO4) という形で以下のようにまとめた*6。
いずれのルールも4にちなんでいることから “Rule-of-Four” と呼ばれている。RO4を満たすような化合物はPPI阻害剤になりやすい性質を有していると考えられる*7。
一方で、RO5とQEDの関係と同様に、定量的に判断できる指標は重要である。我々はQEDの考え方を参考にして、記述子の統計値をもとに「PPI標的化合物らしさ」を定量化できる指標としてQEPPI (https://github.com/ohuelab/QEPPI) を開発した*8。QEPPIでは、関数モデリングに先立ち、Morelliらが用いていたデータよりもより広範なデータを収集するため、論文等からPPI標的化合物を登録しているiPPI-DBを用い、ここから冗長性を省いた1,007化合物を選択した。これらのデータについてQED記述子のうちALERTSを除く7つの記述子の分布*9を求め、QEDと同様に関数モデリングを行って0から1の値としてPPI標的化合物らしさを定量化した。図3にiPPI-DB化合物に対するそれぞれの記述子のヒストグラムを描画し、分布関数を重ねたものを示す。QEDと比較して分布の山がいずれの記述子でも値が大きい方にずれていることがわかるかと思う。
QEPPIの具体的な例として、たとえばコロナウイルスのスパイクタンパク質とヒト細胞表面のACE2タンパク質との相互作用を阻害することが知られている抗ウイルス薬ウミフェノビル (Umifenovir) は、QED = 0.376と低めのQED値を示す一方で、QEPPI = 0.869と高いQEPPI値を示す(図4)。また、実際に開発されている臨床試験段階のPPI標的化合物についてQEPPI値を計算したところ、比較的高い値を示すこともわかった(図5)。
なお、それぞれの記述子の計算にはRDKit version 2022.9.1を用い、QEPPIは以下のPythonコードを用いて計算した。
from rdkit import Chem import QEPPI as ppi m = Chem.MolFromSmiles(r'CCOC(=O)C1=C(N(C2=CC(=C(C(=C21)CN(C)C)O)Br)C)CSC3=CC=CC=C3') q = ppi.QEPPI_Calculator() q.read() q.qeppi(m) # QEPPI #Chem.rdMolDescriptors.CalcNumRings(m) # * RING の計算は CalcNumRings()



タンパク質間相互作用向け化合物ライブラリー
PPIライブラリー
PPI標的薬の設計には、標準的な化合物ライブラリーではなく、PPI標的薬になりそうな化合物を集めたフォーカストライブラリーであるPPIライブラリーを用いる方が、ヒット化合物を得られる可能性が高くなると考えられる。単純に化合物ライブラリーからRO4を満たす化合物を選んで使うだけでも簡易的なPPIライブラリーとして活用できるが、特にタンパク質の相互作用面の特徴などから提案されたPPIライブラリーが、化合物サプライヤーから提供されている。以下に代表的なPPIライブラリーを4つ紹介する。
Enamine社が提供するPPIライブラリー (https://enamine.net/compound-libraries/targeted-libraries/ppi-library) はその代表例である。Enamine社のPPIライブラリーは、複数のタンパク質複合体構造を解析し、PPIに特徴的な二次構造やモチーフに特化して設計された化合物を集めたものであり、さらに忌避構造を含まないようにフィルタリングされ、実際に合成サンプルとして得られている化合物を約4万件収載している。
ChemDiv社が提供するPPIライブラリー (https://www.chemdiv.com/catalog/focused-and-targeted-libraries/protein-protein-interaction-library/) は、ペプチドミメティクスによる化合物や大環状型化合物、スピロ化合物などの特殊な母核構造も含むライブラリーであり、約21万件の化合物からなる。
UkrOrgSyntez社が提供するPPIライブラリー (https://uorsy.com/fragments-and-targeted-libraries/uorsy-ppi-modulators/) では、RO4を満たす化合物であり、PPI相互作用面におけるホットスポット残基であるTyr/Trp/Arg残基との相互作用が期待できる官能基を含む母核が含まれた、約6,500件の化合物からなる。
Life Chemicals社は、複数の方法で構築した多様なPPIライブラリーを提供する (https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/ppi-libraries/)。実際にいくつかのPPIの相互作用面に対してドッキングシミュレーションを行うことによって構築したライブラリーや、RO4によるフィルタリングを行ったもの、公開されているPPI阻害アッセイで阻害能が確認された化合物と類似する化合物を集めたもの、独自に構築した機械学習による分類器で弁別したPPI標的化合物を集めたものなどが含まれている。
以上は化合物サプライヤーが提供するものであるが、公共データベースと論文情報から収集された既知のPPI阻害剤を含むPPIライブラリーとして、慶應義塾大学とペプチドリーム株式会社が共同で開発しているDLiP (https://skb-insilico.com/dlip) がある。DLiPでは、約15,000件のPPI阻害が期待できる化合物と、約26,000件の既知PPI阻害化合物の情報が収載されており、ウェブインターフェース上で検索や詳細閲覧が可能となっている。
PPIバーチャルライブラリー
低分子化合物の既存のライブラリーでも1億件以上の化合物が収載されている(例:ZINC15 in-stock library)が、化合物空間の多様性を考えれば化合物は無数に存在する*11。もちろんそのような化合物の中には合成が困難であったり創薬に適さない化合物も多々含まれるが、検討に値する化合物も大量に存在すると考えられる。そのような、これまでに合成されていないような化合物を計算によって「バーチャルに」生成することをde novo分子設計と呼び、機械学習の発展によって様々なde novo分子設計法が提案され、バーチャル化合物が生成されてきた。
de novo分子設計法の1つであるREINVENT*12は、リカレントニューラルネットワーク (RNN) に基づくSMILES文字列の生成モデルである。事前にChEMBLデータベースの化合物で学習したSMILES生成のための事前モデルから、あらかじめ決められた評価値が良くなる方向に学習を進める強化学習ステップによって、より良い評価値を持つ仮想的な化合物を生成するしくみとなっている。図6に我々がREINVENTを使ってQED、RO4(1ルールを満たすごとに+0.25として設計)、QEPPIの各値を評価値として与えて強化学習を行い、分子生成を行った結果を示す。QEDを良くするように設計されたバーチャル化合物は、当然ながら比較的分子量が小さく、LogPも低い傾向にある。一方でRO4を満たすように設計されたバーチャル化合物は、RO4が下限のみを定めるルールであることから分子量およびLogPが際限なく大きくなってしまうという問題がある。QEPPIでは分布から望ましい記述子の値を取るように設計されるため、QEDよりもやや分子量・LogPが大きい分子が生成されるが、たとえば分子量が1000を超えるような分子は生成されないことがわかる。我々はこのQEPPIに基づいてREINVENTで生成した化合物群を集めて、PPIバーチャルライブラリーとして公開する予定である。

まとめ
この記事では、タンパク質間相互作用を標的とする低分子設計を目指したいくつかの計算手法について紹介した。低分子化合物の創薬に役立つdry手法は、AI技術の発展とともに様々な面で発展し続けており、この記事で紹介した技術はほんの一端でしかない。タンパク質間相互作用はもはや狙えない標的ではないはずなので、これからも多くの薬が誕生することを願ってやまない。
謝辞
本記事の内容について、叢雲くすり氏 (twitter id @souyakuchan) より助言を頂きました。ここに感謝致します。
*1:BioGRIDデータベースから、生物種: Homo sapiens、Experiment type: PHYSICAL、Non-Redundant Interactions(異なる実験手法に基づく同一PPIの確認を単一カウントとする)によって検索した件数。(BioGRID version 4.4.215, 2022年11月現在)
*2:Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov, 15(8), 533-550, 2016.
*3:Thanos CD, DeLano WL, Wells JA. Hot-spot mimicry of a cytokine receptor by a small molecule. Proc Natl Acad Sci U S A, 103(42), 15422-15427, 2006.
*4:Lipinski CA. Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev, 23(1-3), 3-25, 1997.
*5:Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem, 4(2), 90–98, 2012.
*6:Morelli X, Bourgeas R, Roche P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I). Curr Opin Chem Biol, 15, 475–481, 2011. なおこの論文で言及されている2P2Iデータベースは、今はアクセスができない(汗)。
*7:Morelliらが解析したPPI阻害剤は論文等で報告されたものに基づいており、一部を除いて臨床試験段階には到達していない。その意味で、RO4が医薬品としての適正を担保するわけではないため注意が必要である(たとえば生物学的利用能は低くなることが予想される)。
*8:Kosugi T, Ohue M. Quantitative Estimate Index for Early-Stage Screening of Compounds Targeting Protein-Protein Interactions. Int J Mol Sci, 22(20), 10925, 2021.
*9:一般に分子量が大きくなるとその分忌避構造としてカウントされる部分構造を保有する確率も上がるため、ALERTSは分子量と相関し、PPI標的化合物では平均的に高い値になると考えられる。しかし、iPPI-DBに収録されている化合物のALERTSの平均値は、経口医薬品のALERTSの平均値よりも低くなるという、直感に反する結果が得られた。このことから、(初期の段階ではALERTSが低い化合物が優先的にアッセイにかけられている可能性など、)iPPI-DBのデータにはALERTSに関する選択的バイアスがあると考え、QEPPIのモデリングからALERTSを除外した。
*10:Truong, J.; George, A.; Holien, J.K. Analysis of physicochemical properties of protein-protein interaction modulators suggests stronger alignment with the “Rule-of-Five”. RSC Med Chem, 12, 1731-1749, 2021.
*11:RO5を満たす薬理活性化合物だけを考えた場合でも、潜在的に個超の化合物が存在すると推定されている。
*12:Blaschke T, Arús-Pous J, Chen H, Margreitter C, Tyrchan C, Engkvist O, Papadopoulos K, Patronov A. REINVENT 2.0: An AI Tool for De Novo Drug Design. J Chem Inf Model, 60(12), 5918-5922, 2020.