この記事は?→創薬 (dry) Advent Calendar 2022の5日目の記事です。 adventar.org
10年以上の年月と3,000億円超の費用を要する医薬品開発の期間とコストを削減するため、創薬の様々な場面でコンピュータによるインシリコ(in silico)解析が導入されている。特に通常の低分子化合物では、バーチャルスクリーニングによるヒット化合物の発見、標的分子と活性リガンドの構造情報を利用した分子設計によるリード化合物の最適化、物性や動態の予測・最適化に至るまで、様々な課題に対して計算論的手法が開発されてきた。かたやペプチドについては、低分子と比較するとやや「出遅れていた」状況であった。だが、近年の技術革新によりインフォマティクス技術を基盤としたペプチドの設計が実現しつつある。以下ではペプチド創薬に活用できる、ペプチドを対象としたインシリコ(in silico)解析技術を紹介する。
タンパク質-ペプチドドッキング
ドッキングシミュレーション(単にドッキングとも言う)は、標的となる分子と目的の分子の複合体構造を推定し、併せて標的親和性を計算によって評価するための方法論である。標的としてはタンパク質やRNA、目的の分子としては低分子や金属イオン、糖鎖などが該当し、たとえばタンパク質と低分子(タンパク質-リガンドドッキング)、タンパク質とタンパク質(タンパク質ドッキング)、タンパク質と金属イオン、タンパク質と糖鎖など、個々に手法が開発されている。このうち、タンパク質とペプチド(主に数残基~20残基程度のサイズ)を対象とした技術をタンパク質-ペプチドドッキングと呼ぶ。
タンパク質-ペプチドドッキングは、標的タンパク質に対するペプチド分子の結合様式(複合体構造)を推定し、その結合親和性をエネルギースコア等の値として評価するような手法を言う。いくつかのソフトウェアが提案されているが、なかでもAutoDock CrankPep*1が、数残基~20残基くらいまでのペプチドに対して精度良く複合体構造(図1)を推定可能であるとされている。

ドッキングソフトウェアから計算されるエネルギースコアの値の良し悪しで、ペプチドの標的タンパク質に対する結合能が評価できる。エネルギースコアは「タンパク質のどこにどんな形で結合しそうか」を評価するための計算値であるが、たとえば「目的のペプチドが、他のすでに知られている結合ペプチドや、ランダムな配列のペプチドと比較して、どのくらい結合能が高い(or低い)のか」を推定すること、すなわちバーチャルスクリーニングにも活用できる。
なお、ほとんどのドッキングソフトウェアでは直鎖かつ標準アミノ酸20種で構成されたペプチドしか扱うことができなかったが、最近ではD体アミノ酸や修飾のあるアミノ酸などの非標準アミノ酸を含んだペプチドや、ペプチド鎖の末端や途中で環状型になっているペプチド(環状ペプチド)についても入力として扱えるソフトウェアが増えてきた。AutoDock CrankPep(環状ペプチドに対する評価は文献*2を参照)やHADDOCK*3は、環状ペプチドに対するドッキングシミュレーションを実行可能なソフトウェアとしても使われている。
ところで、タンパク質立体構造予測の分野ではAlphaFold2による予測精度の革新があったことは記憶に新しい。実は現在のAlphaFold2は、タンパク質複合体(ホモ/ヘテロオリゴマー)の構造予測が可能となっており、タンパク質ペプチド複合体構造の予測にも使えそう(図2)だということが複数のグループによって検証されてきた*4*5*6。

世界で初めてAlphaFold2によるペプチドドッキングを報告したツイート。残念ながら後の論文からはreferされず。(英語でtweetしておけばよかった)あ、AlphaFold2でペプチドドッキングできちゃった pic.twitter.com/BkNs6davJR
— Ohue M/大上雅史 (@tonets) 2021年7月20日
特にこれらの報告の中で、従来のドッキングと同様にペプチドのバーチャルスクリーニングができる可能性が示唆されている。ただし、AlphaFold2の制限として直鎖・標準アミノ酸のペプチドしか扱うことはできないことに注意が必要である。さらなる応用として、たとえば2つのペプチド配列を入力して、より結合が強い方がタンパク質に結合するような結果が得られるという、競合ドッキング法(competitive binding)と呼ばれる方法も提案されている*7(図3)。
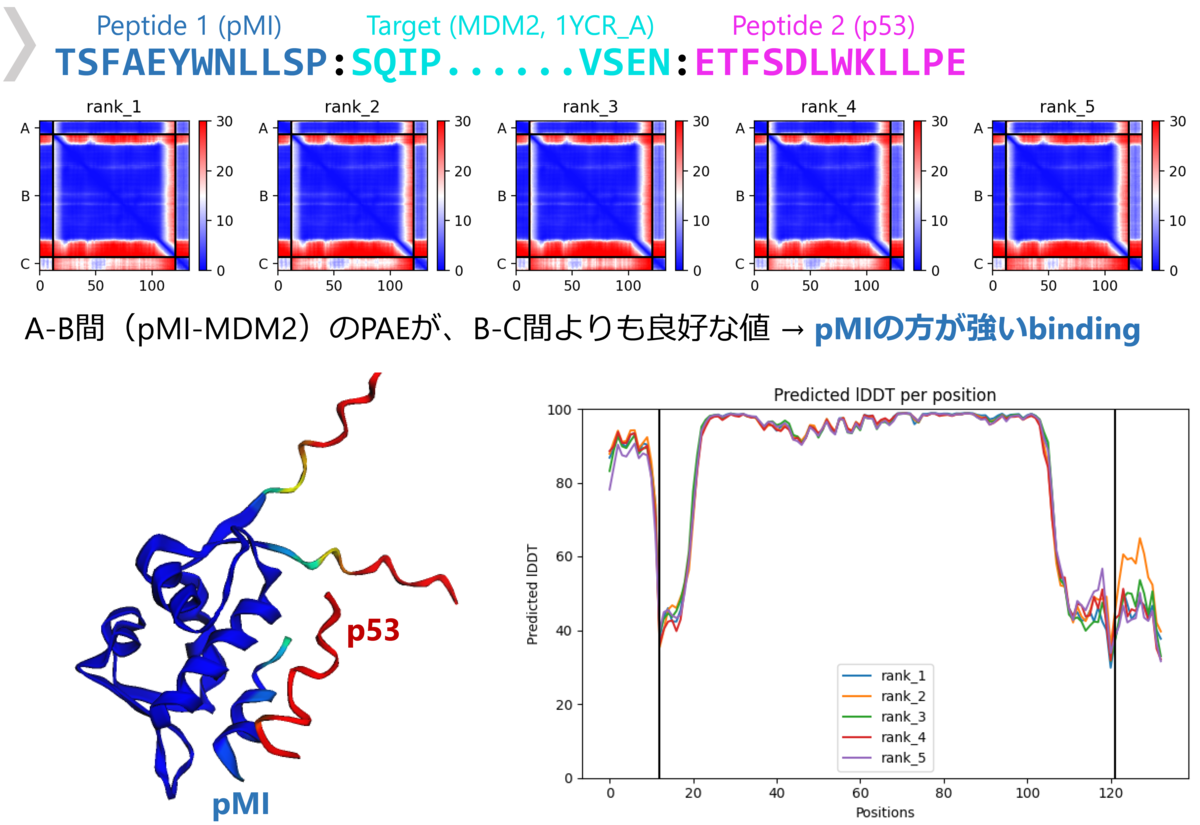
AlphaFold2によるペプチドデザイン
計算によって標的タンパク質に対するペプチドの結合能を評価することができるということは、手元にあるペプチド配列の比較だけでなく、あらゆるペプチド配列をソフトウェアに入力していくことで、標的タンパク質に対して結合能が高いペプチド配列が見つけられるということになる。ただしこのような発想で実際に網羅的にペプチド配列のスクリーニングを行う場合には、ペプチド残基長nに対して20ⁿ回の計算をする必要がある。10残基のペプチドだけを考えたとしても20¹⁰ ≒ 10兆通りのペプチド配列の評価が必要になり、これは非現実的である。対して、ランダムな配列からスタートし、予測の結果と欲しい構造との差を勾配としてフィードバックにして、フィードバックの結果から次の配列を選んでいくという方法(Hallucination法)*8が提案されており、予測値が良くなる方向にバイアスをかけて配列を選択することで、全通りの計算を避けて妥当なペプチド配列を生成することが可能になっている。
このアイデアに基づいて、Hallucination論文やRoseTTAFoldの著者でもあるSergey Ovchinnikovらによって実装されたAfDesignのbinder hallucination法で、実際にペプチド配列が生成できるようになっている。AlphaFold2の出力するペプチド複合体予測の評価値(pLDDTやPAEなど)を良くする方向にペプチド配列をサンプリングしていくことで、AlphaFold2的に良いと考えるペプチドを現実的な計算時間で生成するという仕組みである。実際にAfDesignを実行すると、実際に図4のようにペプチド配列が決定されていく様子を動画で出力することができる。

Adding support for binder hallucination if anyone wants to try! (Code is very experimental, not intended for practical use... only use for art/science) 😀https://t.co/OOPp8kSu2Z pic.twitter.com/WUu9LGKIwp
— Sergey Ovchinnikov @ NeurIPS 🇺🇦 (@sokrypton) 2022年2月3日
AlphaFold2による水溶性ペプチドデザイン
ところが、AfDesignによって生成された配列を確認すると、難水溶性のペプチドが多くを占めていた。タンパク質間相互作用の相互作用面は一般に疎水領域であり、AlphaFold2 (AlphaFold-Multimer) も相互作用面に共起しやすい残基の関係を学習していると考えられることから、タンパク質の表面に結合するペプチド配列をAlphaFold2によって設計しようとすると疎水領域を構成するように、すなわち疎水性残基を多用したペプチド配列が選ばれやすくなっているのだと解釈できる。だが、後の生化学実験などを考える上ではペプチドの水溶性は重要である。
我々はこの問題を解決するために、AlphaFold2の評価値に加えてhydropathy indexなどのアミノ酸に関する物性評価指標を導入し、「疎水性アミノ酸はあまり使わないように」といった形で残基の使用頻度を制御することで、適切なペプチド配列を生成する手法「Solubility-AfDesign」を提案した*9。Solubility-AfDesignによって、実際に標的結合能を維持したまま水溶性を向上させるペプチド配列の予測に成功し(図5)、具体的な複合体構造モデルとともに提示することができるようになった。この論文は日本語で以下に解説があるので、興味を持った方は読んで頂ければ幸いである。 blacktanktop.hatenablog.com
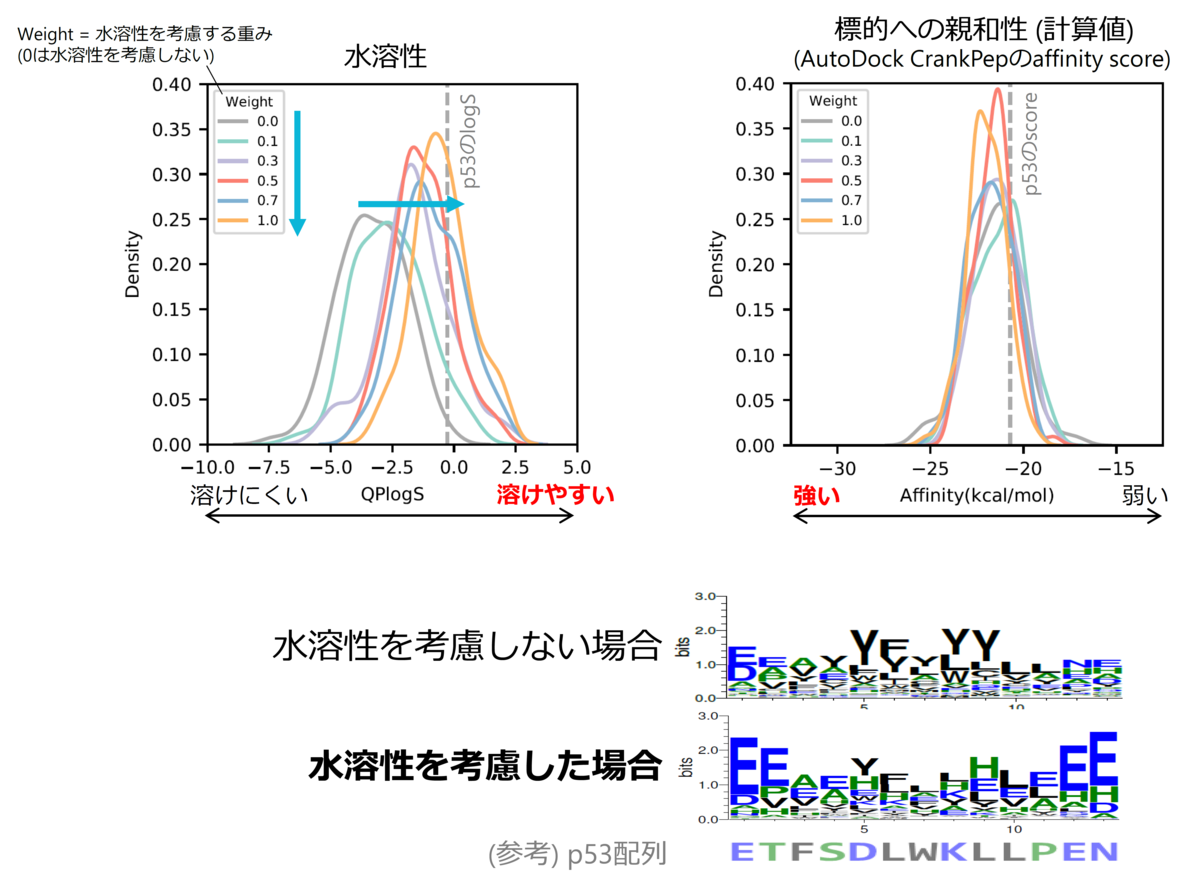
まとめ
本稿ではペプチドに関するin silico技術を紹介したが、ペプチドの計算技術・設計技術はまだまだ発展を続けている段階である。タンパク質構造、低分子、ペプチドと、それぞれでAIやシミュレーション技術が展開されてはいるが、まだまだin silicoが追いついていない領域でもあるかと思う。AlphaFold2やChromaのように突然すごいAI技術が降ってくるかもしれない。どれがイケててどれは微妙なのか、目利きも重要になるかと思う。今後の発展に目が離せない。
Today we introduced Chroma, a generative model that creates new proteins & protein complexes given geometric & functional constraints. It learns to transform unstructured, random 3D shapes into #protein molecules, which can have tens of thousands of atoms. https://t.co/cORRnRKnfB pic.twitter.com/2OUk9AOQuj
— Generate Biomedicines (@generate_biomed) 2022年12月1日
*1:Zhang Y, Sanner MF. AutoDock CrankPep: combining folding and docking to predict protein-peptide complexes. Bioinformatics. 2019;35(24):5121-5127. doi: 10.1093/bioinformatics/btz459.
*2:Zhang Y, Sanner MF. Docking Flexible Cyclic Peptides with AutoDock CrankPep. J Chem Theory Comput. 2019;15(10):5161-5168.
*3:Charitou V, van Keulen SC, Bonvin AMJJ. Cyclization and Docking Protocol for Cyclic Peptide-Protein Modeling Using HADDOCK2.4. J Chem Theory Comput. 2022; 18(6):4027-4040.
*4:Ko J, Lee J. Can AlphaFold2 Predict Protein-Peptide Complex Structures Accurately? bioRxiv 2021.07.27.453972, 2021.
*5:Tsaban T, Varga JK, Avraham O, Ben-Aharon Z, Khramushin A, Schueler-Furman O. Harnessing protein folding neural networks for peptide-protein docking. Nat Commun. 13(1):176, 2022.
*6:Johansson-Åkhe I, Wallner B. Improving peptide-protein docking with AlphaFold-Multimer using forced sampling. Front Bioinform, 2, 959160, 2022.
*7:Chang L, Perez A. AlphaFold encodes the principles to identify high affinity peptide binders, bioRxiv 2022.03.18.484931, 2022.
*8:Anishchenko I, Pellock SJ, Chidyausiku TM, Ramelot TA, Ovchinnikov S, Hao J, Bafna K, Norn C, Kang A, Bera AK, DiMaio F, Carter L, Chow CM, Montelione GT, Baker D. De novo protein design by deep network hallucination. Nature. 2021; 600(7889):547-552.
*9:Kosugi T, Ohue M. Solubility-aware protein binding peptide design using AlphaFold. Biomedicines, 10(7): 1626, 2022.